

 Click it and Unblock the Notifications
Click it and Unblock the Notifications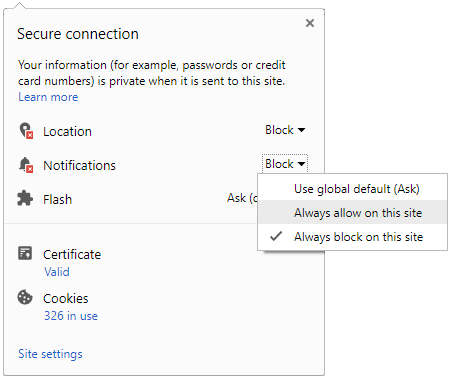

Just In



Don't Miss
-
 Heeramandi First Review: Sanjay Leela Bhansali’s Period Drama Is Special & Mesmerising; Deets Inside
Heeramandi First Review: Sanjay Leela Bhansali’s Period Drama Is Special & Mesmerising; Deets Inside -
 Global NCAP Lauds Tata Motors - A 5-Star Journey
Global NCAP Lauds Tata Motors - A 5-Star Journey -
 Gold Prices Ayodhya: In Ram-Janma-Bhumi, 24carat Falls By Rs 380 In 10-Grams, Drops By Rs 3,800 In 100-gram
Gold Prices Ayodhya: In Ram-Janma-Bhumi, 24carat Falls By Rs 380 In 10-Grams, Drops By Rs 3,800 In 100-gram -
 SRH vs RCB My11Circle Prediction IPL 2024 Match 41: HYD vs BLR Fantasy Tips & Expert Picks
SRH vs RCB My11Circle Prediction IPL 2024 Match 41: HYD vs BLR Fantasy Tips & Expert Picks -
 JEE Main Result 2024 Out, Check Category- Wise Toppers' List Here
JEE Main Result 2024 Out, Check Category- Wise Toppers' List Here -
 Heeramandi Screening: Alia Bhatt, Ananya Panday, Rashmika Mandanna And Others Serve Finest Ethnic Style!
Heeramandi Screening: Alia Bhatt, Ananya Panday, Rashmika Mandanna And Others Serve Finest Ethnic Style! -
 Qubo InstaView Video Door Phone Launched in India: Check Price, Features
Qubo InstaView Video Door Phone Launched in India: Check Price, Features -
 Escape to Kalimpong, Gangtok, and Darjeeling with IRCTC's Tour Package; Check Itinerary
Escape to Kalimpong, Gangtok, and Darjeeling with IRCTC's Tour Package; Check Itinerary

Counting duplicated genome segments now possible with new computational method

London, August 31 (ANI): Counting copies of duplicated genome sequences and doing initial analyses of their contents are possible with the aid of a new computational method, according to a study.
Led by scientists at the University of Washington (UW), the study suggests that the number of copies of particular DNA segments can differ from one person to the next.
The researchers use the term mrFAST, an acronym for micro-read Fast Alignment Search Tool, to refer to the novel method.
In their study report, they have highlighted the fact that segmental duplications in the human genome have been associated with susceptibility and resistance to disease.
The report points out that duplicated segments have been linked to such disorders as lupus, Crohn's disease, mental retardation, schizophrenia, colour blindness, psoriasis, and age-related macular degeneration.
It adds that segmental duplications often contain duplicated genes, many of which have an unknown function, and that individuals have different numbers of copies of some of these duplications.
The researchers write that determining the number, content, and location of segmental duplications is an important step in understanding the health significance of gene copy-number variation.
"New computational methods, combined with next-generation DNA sequencing technology, has provided for the first time an accurate census of specific genes that exist in varying number of copies," Nature magazine quoted Alkan as saying.
"This is a way to deal with some of the most complex regions of the human genome and do what might appear to be a simple thing: Count whether a person has one, two, three or more copies of a gene. In fact, such counting is surprisingly difficult," said Kidd.
The researchers say that next-generation technology for sequencing the human genome has far greater detection power, and costs substantially less than the traditional sequencing method known as Sanger sequencing.
According to them, the new technologies are beginning to distinguish subtle dissimilarities between nearly identical gene copies.
"This can provide researchers with a more accurate assessment of specific gene content and insight into functional constraints," Alkan said.
"The newer, faster genome sequencing platforms may eventually make it feasible to detect the full-spectrum of genomic variation among many individuals, including patients suffering from diseases of genetic origin. Next-generation technology and computational methods promise low cost, rapid sequencing of different individuals and may lead to a fuller understanding of the patterns and significance of human genetic variation," Alkan added.
The analytical method they devised is already being tapped for the 1000 Genome Project, an international effort to catalog and compare the genomes of hundreds of people from around the world.
Alkan, Kidd, and their colleagues note that the ability to accurately and systematically determine the absolute copy number for any genomic segment is a notable step toward a true and complete picture of individual genomes, and how the genome shapes a person's characteristics.
"The next challenge will be defining variation in the sequence content and the structural organization of these dynamic and important regions of the human genome," they wrote.
A research article describing their study has been published in the journal Nature Genetics. (ANI)


